AIP is a disease characterized by peculiar pathologic findings (fibrosis and lymphocyte infiltration), laboratory data (increased serum IgG levels and autoantibody), and effectiveness of
corticosteroid therapy. AIP shows a male preponderance and affects usually patients in fifth-seventh decades. Association with other autoimmune diseases (for example Grave’s disease) has been
reported. Pathologically, AIP is classified in diffuse and focal type, which usually involves the pancreatic head. Lymphocytic inflammatory infiltrate, fibrotic changes and variable degree of
parenchymal atrophy are the main histologic features. Patients may present with obstructive jaundice, mild upper abdominal pain, weight loss and easy fatigability. Laboratory examinations may show an
increased levels of IgG and/or IgG4 and the presence of autoantibodies (antinuclear, anti-carbonic anhydrase antibodies, etc.) pointing out the autoimmune origin of this disease. US examination
depicts homogeneous, low echogenic, diffuse or focal enlargement of the pancreas, sometimes associated with dilation of the intrahepatic and common bile ducts. After US contrast-media administration,
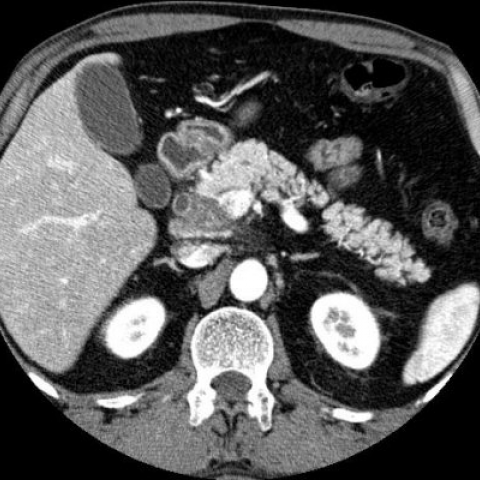
the involved pancreatic parenchyma shows from mild to moderate hyperechoic enhancement, related to the grade of inflammatory involvement and its fibrotic evolution. On CT, the diffuse type of AIP
appears as a uniform swelling of the pancreas with well-defined outline. Sometimes it can be observed an hypodense rim-like capsule surrounding the pancreas showing a delayed post-contrast
enhancement (related to fibro-inflammatory changes involving peripancreatic adipose tissue). In the focal type, CT examination depicts a mass-like lesion located in the head and/or in the uncinate
process. Before contrast-medium administration, this focal enlargement typically has smooth margins and it appears homogeneous and iso-hypoattenuating respect the surrounding pancreas. On
contrast-enhanced images the lesion usually shows a hypodense appearance in pancreatic phases and become isodense or slightly hypodense in the venous phase. Dilation of the main pancreatic duct and
biliary tree may be depicted. Parenchymal calcifications, pseudocysts, vascular invasion, inflammatory involvement of the mesentery, the anterior pararenal fascia and the lateroconal fascia are
typically absent. MR examination demonstrates a homogeneous, focal/diffuse pancreatic enlargement, generally hyper-isointense in T2-weighted images and hypointense in T1-weighted sequences compared
to the liver. In gadolinium-enhanced T1-weighted images, the involved pancreatic parenchyma shows a homogeneous enhancement. A capsule-like rim surrounding the pancreas appears as a hypointense
peripheral band on both T1 and T2-weighted images and may show delayed enhancement. MRCP and ERCP often show diffuse/focal narrowing of the pancreatic duct. In the absence of supporting clinical
findings and serologic marker levels, a focal form of autoimmune pancreatitis may be extremely difficult to differentiate from pancreatic cancer on the basis of imaging features alone. Imaging
features that can help in differentianting autoimmune pancreatitis from pancreatic cancer include homogeneous enhancement of the involved pancreas, absence of substantial pancreatic duct dilatation
and parenchymal atrophy, and the absence of vascular encasement or metastatic disease. Concomitant diffuse pancreatic ductal narrowing and common bile duct/intrahepatic biliary strictures that
resemble primary sclerosing cholangitis should also raise the probability of a diagnosis of autoimmune pancreatitis. Corticosteroid therapy can determine resolution of clinical manifestation,
normalization of laboratory alterations, and complete or partial regression of pancreatic abnormalities.